
How to give a twist to the self-assembly of nanoparticles
über
In nature, molecular self-assembly plays an important role. For example, proteins stick to each other to form viruses, or protein complexes that transmit signals in cellular networks. Such processes are a source of inspiration for chemists who design the molecular building blocks for functional materials arising through spontaneous self-assembly. Here, in addition to proteins, also synthetic polypeptides or colloidal particles are used. Through the structure of such blocks one can influence the process of self-assembly, and thus the functionality of the final material.
Process dynamics is important
In order to make materials with special properties, scientists would like to predict the outcome of the self-assembly. Typically, they calculate the most stable material structure based on the predicted interaction between the individual building blocks. However, the end result also depends on the dynamics and kinetics of the self-assembly process.
In collaboration with colleagues of Utrecht University, Willem Kegel en Jan Groenewold, ACMM researchers Arthur Newton and Peter Bolhuis have now studied the self-assembly kinetics using computer simulations for a simple model of proteins. They focused on the influence of the rotation of the particles in the self-assembly. Their work was financed by the FOM Foundation.
Rotational diffusion not always in accordance with Einstein
In many self-assembly-simulations, the rotation of particles is considered in conjunction with their translation. Both aspects are usually linked via the so-called Stokes-Einstein relation. However, in recent years, experiments have revealed that this coupling does not always hold. This applies in particular under conditions which are of interest in self-assembly, such as in tightly packed, crowded environments like the cytoplasm of the living cell.
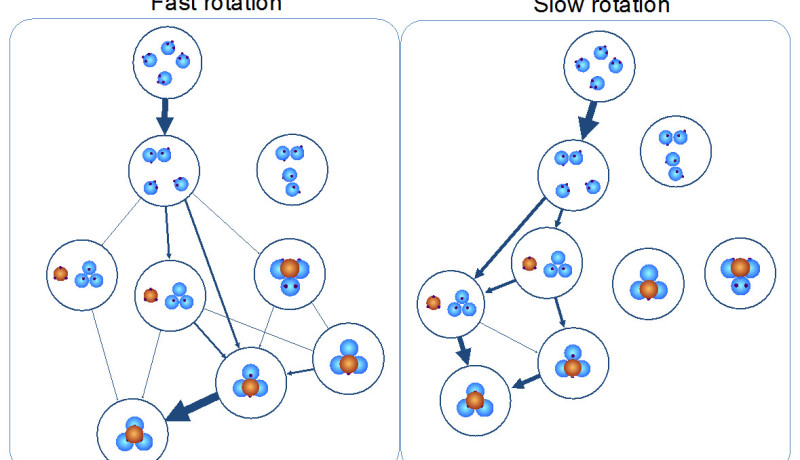
With their simulations, the Amsterdam researchers investigated the result of releasing the Stokes-Einstein coupling. Even for two particles already a strong effect on the association emerges. The effect is even more relevant for systems with multiple particles, when metastable intermediate states are possible. In a simulation of cluster formation of four particles, varying the rotational diffusion significantly shifts the preference for the self-assembly routes. The researchers were able to generalise this finding to clusters of any size.
New materials
The researchers argue that their results provide new opportunities for a better control of the bottom-up synthesis of functional materials. For example, it should be possible to design particles of which the rotation can be controlled with magnetic or electric fields.
The study also gives an impetus to better understanding of the influence of rotational diffusion on self-assembly processes in nature, especially in very crowded environments such as molecular in the cytoplasm of a living cell. Amongst others, this discovery will have consequences for the modeling of protein complex formation, such as in viruses.
Article
Arthur C. Newton, Jan Groenewold, Willem K. Kegel and Peter G. Bolhuis: Rotational diffusion affects the dynamical self-assembly pathways of patchy particles. PNAS Early Edition, published online 30 November 2015, DOI: 10.1073/pnas.1513210112.
Diskussion (0 Kommentare)